Hiperconjugação
O termo hiperconjugação, também conhecido como efeito Baker-Nathan, foi utilizado pela primeira vez por John W. Baker e W. S. Nathan em 1935.. Estes pesquisadores utilizaram o termo para explicar a lei da velocidade de reação em uma série de reações entre piridina e brometo de benzila substituído com grupos alquila na posição 4. Como os resultados não podiam ser explicados pelo efeito de campo, o novo conceito foi concebido e, a partir de 1940, com Robert S. Mulliken passou a ser amplamente utilizado.
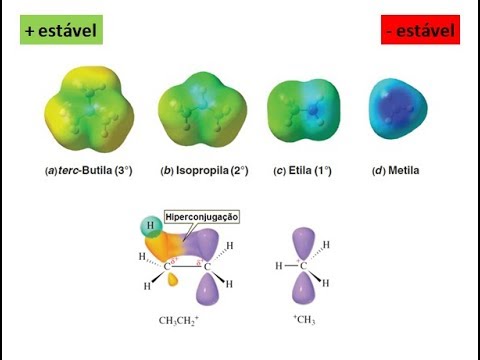
Hiperconjugação é uma das maneiras que existem para descrever a deslocalização eletrônica nas moléculas. A mais conhecida e que tem grande importância na química orgânica ao longo de muitos anos é a conjugação. A conjugação se refere à deslocalização de elétrons Π em sistemas conjugados. Enquanto o termo hiperconjugação se refere à deslocalização de ligações sigmas. A hiperconjugação pode ser uma resposta a estudos teóricos e experimentais que evidenciam significativas mudanças na geometria, na densidade de elétrons, em espectros de infravermelho (IR), e propriedades em ressonância magnética nuclear (RMN), podendo ainda influênciar em equilíbrios conformacionais, determinando a seletividade e as energias do orbital molecular (MO). Segundo a teoria dos orbitais moleculares (MO), hiperconjugação é descrita como a interação entre orbitais eletrônicos, onde um deles corresponde a ligação sigma. O fenômeno é praticamente o mesmo que a conjugação, porém a interação tem que estar estabilizando um sistema, onde o orbital de mais alta energia tem que estar vazio ou parcialmente vazio (1 elétron), enquanto o outro de menor energia tem que estar parcialmente preenchido (Figuras 2 e 3).
A hiperconjugação pode ser classificada de diferentes formas dependendo do número relativo de elétrons que contribui para a estabilização da ligação. Mulliken foi quem iniciou com estas classificações, e se referiu a hiperconjugação de duas maneiras: sacrificial (ordinária ou heterovalente) para sistemas neutros e isovalente (ou forte) para sistemas catiônicos, tendo ainda a possibilidade de ser para sistemas anionicos. A Hiperconjugação positiva domina quando sistemas com orbitais p ou sistemas π fortemente aceptores ou sistemas sigmas fortemente doadores estão presentes na molécula. Isto é comum em cátions ou também moléculas neutras como boranas. Um exemplo de efeito estabilizador por hiperconjugação positiva pode ser vista em estudos de β-sililetil cátions, que apresentam estabilidade em torno de 38 Kcal/mol em fase gasosa. Já a Hiperconjugação Negativa foi contactada em ligações C-F ao invés de ligações C-H, onde a polaridade se inverte.
Assim como a conjugação, que é vista normalmente como formas de “comunicação” a distância entre orbitais que estejam conjugados, ou seja, como “pontes” para transportar elétrons dentro da molécula, a hiperconjugação, embora menos esclarecida, apresenta alguns relatos de deslocalização estendida, denominadas de homohiperconjugação e dupla hiperconjugação. A homohiperconjugação é vista quando o centro insaturado entre aceptores e doadores de elétrons se invertem, ou quando o centro aceptor é um orbital p catiônico mais afastado do centro doador, ao se comparar com a cojugação. Já a dupla hiperconjugação amplia mais a deslocalização eletrônica quando existe uma ponte sigma entre o doador e o aceptor, sendo geralmente um orbital catiônico.
Lambert e Singer realizaram uma série de experimentos baseando-se nas constantes de acoplamento de ligação C-C para explicar a hiperconjugação sacrificial. Tal experimento consistia na obtenção de uma série de substâncias variando grupos doadores (M) e retiradores (X). Onde, para X = H, Me e MeO, substratos foram preparados para todos os átomos M. Para X = CN, apenas M = Si e Sn foram preparados, e para X = NO2 apenas M = C foi preparado. Através desse experimento eles observaram, como esperado, que a hiperconjugação aumenta a ordem de ligação entre os carbonos benzílicos e ipso (CH2-Ci) e diminui a ordem de ligação entre o átomo doador e o carbono benzílicos (CH2-M) e em termos de estrutura, o comprimento de ligação CH2-Ci diminui e aumenta a constante de acoplamento e para comprimento de ligação CH2-M ocorre o inverso. Deve-se contudo salientar que as constantes de acoplamento C-C dentro do anel aromático são essencialmente independentes das mudanças no M. Dessa forma, os fatos experimentais vão contra a hipótese original de Baker-Natan, visto que a hiperconjugação não resulta em alternância de títulos de comprimento dentro do anel aromático.
Overlap e simetria orbitalar
Para que se tenha uma boa interação, os orbitais vizinhos devem ser coplanares, e ainda, estudos tem demonstrado que o arranjo geométrico antiperiplanar é preferencial frente ao sinperiplanar. Este efeito é evidenciado, por exemplo, na maior estabilidade da conformação trans do butadieno frente a conformação cis. As característi as conformacionais, estruturais e reacionais tem grande interferência do efeito de hiperconjugação. Um ponto importante das interações que envolvem hiperconjugação é a sua componete estereoeletrônica. Tais interações dependem da sobreposição de orbitais (formando, preferencialmente, ligações sigmas). As forças que controlam a barreira de rotação em torno das ligaçãos são usadas comumente para a análise conformacional de uma dada estrutura. O etano é um dos sistemas mais utilizados em estudos dessa natureza. A menor energia da estrutura alternada frente a estrutura eclipsada do eteno (cerca de 3 kcal/mol para essa barreira de rotação), tem sido, geralmente, atribuído a estéreo repulsão entre os elétrons da ligação C-H . Mulliken, já em 1939, supôs que a hiperconjugação desempenha um papel fundamental no potencial de rotação tanto do etano, como de outras moléculas .
Barreira rotacional: Etano
A busca para determinar e interpretar a barreira rotacional interna das moléculas foi inicialmente estudado por Pitzer e Lipscomb (LP) Eles estudaram a molécula do etano, nas conformações escalonadas(S) e eclipsada (E) (figura 18) considerando que o comprimento de ligação e ângulos de ligação nessas conformações são iguais. O baixo valor para a barreira rotacional do etano, 3,3 kcal/mol foi atribuído ao efeito estérico de repulsão entre os elétrons na ligação C-H na conformação de eclipse. A rotação, que causa o enfraquecimento da ligação C-C e a hiperconjugação, são os fatores responsáveis para a alta estabilidade na conformação escalonada. Segundo Pophristic e Goodman usando análise Natural bond orbitals (NBO), encontraram que a conformação em forma de eclipse é a estrutura majoritária.. Esse estudo entretanto foi contestado recentemente , mostrando que há um artefato nos cálculos NBO.
Existem controvérsias a respeito da hiperconjugação neutra, porém alguns trabalhos baseados em dados termodinâmicos ressaltam a importância da hiperconjugação em alcenos e alcinos. Roger et al observou que a estabilidade atribuída a conjugação π do composto 1,3 butadiino é praticamente zero quando estimado através da abordagem clássica de Kistiakowsky et al. Kistiakowsky et al sugeriu que a estabilidade do 1,3 butadieno pode ser avaliada através da liberação energética da reação de hidrogenação do composto como mostra o esquema ao lado: De acordo com Kistiakowsky et al a reação de hidrogenação do 1,3 butadieno a 1-buteno é menos energética que a reação de hidrogenação do 1-buteno a butano. Kistiakowsky et al associa esse fato a maior força da conjugação π no composto 1,3 butadieno. Já para 1,3- Butadiino foi observado que a liberação energética nas duas etapas de hidrogenação (ver esquema) foi igualmente exotérmica.
Equilíbrio conformacional
Pode-se citar ainda como exemplo da importância da hiperconjugação neutra em propeno (pode ser fornecido por seu perfil conformacional). Para o propeno a conformação mais estável é o “eclipsado”, pois uma ligação metil C-H eclipsa a ligação com o carbono vizinho σC-C. A conformação escalonada em uma ligação metil C-H eclipsa os carbonos vinil adjacentes da ligação C-H, é menos estável por aproximadamente 2 kcal/mol. Estas nomenclaturas são inadequadas pois a conformação“eclipsada” do propeno é estereoeletronicamente semelhante à conformação escalonada do etano (figura 18) e vice-versa. Lin et al. comprovaram por análises NBO (análises dos orbitais naturais de ligações) que a hiperconjugação é a mais importante interação vista entre os grupos metil e vinil e divide-se em três partes: a interação C=C πCH3 → π*, a interação C=C π* CH3 → πC e as interações entre orbitais vizinhos no plano σC-H do grupo metil e σ*- orbital do vinil antiperiplanar da ligação C-H. Um esclarecimento análogo foi apresentado por Basso et. al., sobre a proveniência das preferências conformacionais em compostos carbonilados.
A capacidade aceptora de ligações sigma C-X em etanos monossubstituídos com relação ao mesmo doador (ligação C-H antiperiplanar) aumenta em direção ao fim de um período e para baixo nas famílias. O aumento da capacidade aceptora de ligações sigma C-X em períodos, paralelamente ao aumento eletronegatividade, como resultado de mudanças favoráveis na polarização s*, aumenta a matriz Fock e elementos de sobreposição da matriz para a interação [65 e 129]. Por outro lado, essa tendência é oposta quando se pensa nas famílias, já que a capacidade aceptora aumenta para baixo, e a eletronegatividade para cima. Essas comportamento pode ser compreendido a partir da equação da análise NBO (Natural Bond Orbital). A ordem relativa de capacidade aceptora (a energia da interação sC-H -> s*C-X é dada entre parênteses) é a seguinte: X = Br (6,3) > Cl (6,2) > SH(1) (5,4) > F (5,1) > OH(1) (4,7) = SH(2) (4,7) = SeH (4,7) > PH2(1) (4,6) > AsH2 (4,5) = NH2(1) (4,5) > OH(2) (4,2) > PH2(2) (4,0) >NH2(2) (3,8) = GeH3 (3,8) > SiH3 (3,6) > CH3 (3,4) > H (3,2). Dois valores para alguns substituintes correspondem a diferentes conformações. Curiosamente, efeitos estereoeletrônicos exibidos por ligações C-X com X pertencente ao segundo e terceiro períodos são altamente anisotrópicas. Ligações C-calcogênio, por exemplo, são excelentes aceptores-s e na ponta referente ao carbono, mas péssimos aceptores-s no terminal do calcogênio.
Propriedades doadoras de pares solitários
As diferenças relativamente sutis nas energias envolvidas na hiperconjugação tornam-se mais pronunciadas e quimicamente significativas em interações envolvendo melhores doadores, como o par solitário do nitrogênio. Os dados NBO de a-halogenoaminas indicam que tanto a alta energia do orbital não-ligante e sua alta polarizabilidade contribuem para uma energia de interação incrementada.
Hibridização em pares solitários
As diferenças em hibridização são particularmente importantes para interações estereoeltrônicas hiperconjugativas por várias razões. Primeiro, a hibridização é diretamente relacionada à geometria molecular, e determina os ângulos e direção nos quais os orbitais não-ligantes são projetados no espaço para a sobreposição com os orbitais aceptores. Segundo, ela controla o tamanho relativo dos dois lobos de um par solitário. Os lobos frontal e posterior são equivalentes para pares puramente p, enquanto o posterior diminui de tamanho com o decréscimo do caráter p no híbrido spn. Terceiro, a hibridização do orbital doador é relacionada a sua energia absoluta. Um incremento no caráter p leva a um aumento na energia do orbital que diminui a diferença energética entre o par solitário doador e um orbital aceptor s* ou p*. Em geral, a capacidade doadora é diretamente proporcional ao caráter p do par solitário. Pares solitários com 100% de caráter p são melhores doadores que doadores hibridizados spn.
Energia dos pares solitários
A energia de interação é inversamente proporcional à diferença de energia, que depende da energia relativa do par solitário. As tendências na energia de pares solitários podem ser prontamente compreendidas em termos de sua hibridização e da eletronegatividade de X. Aumento na eletronegatividade e diminuição no caráter p abaixam a energia orbital do par solitário. Embora o oxigênio seja mais eletronegativo que o nitrogênio, a hibridização puramente p axial deste, comparada à do nitrogênio, tem um efeito menor. Nesse caso, o efeito da hibridização compensa a diferença de eletronegatividade.
Segundo Cramer , cálculos em fase gasosa são consistentes com uma grande deslocalização por hiperconjugação da densidade do par de elétrons isolados do Oxigênio do anel de Tetrahidropirano no orbital axial da ligação C-N(+)σ*. Os papéis relativos aos efeitos homoanoméricos W- e Plough em aza-, oxa-, tio e selenoheterociclos foram investigados em experimentos com RMN13C e análises NBO, e revelaram que tais efeitos desempenham um importante papel na tendência relativa na constante de acoplamento de uma ligação 1J(C-H), necessária no entendimento das propriedades conformacionais de carbohidratos, azacarbohidratos e outros substratos de interesse biológico . Embora os efeitos homoanoméricos sejam considerados mais fracos que as clássicas interações vicinais anoméricas, sua importância aumenta significantemente quando a habilidade aceptora dos orbitais σ* aumentam em consequência do alongamento da ligação e/ou polarização. Por exemplo, a solvólise de piperidinas e pirrolidinas com um grupo de saída no Carbono β se dá através da formação cátions aziridínicos cíclicos, devido a ajuda anquimérica do par isolado do Nitrogênio .
Histórico
Originalmente, define-se efeito anomérico como a tendência de heteroátomos substituintes, adjacentes a um heteroátomo formador de um ciclohexano saturado cíclico, em assumir uma orientação axial ao invés de uma orientação equatorial, menos impedida estericamente. Esse termo foi utilizado na literatura porque envolve o Carbono 1 de derivados de piranoses e de glicopiranosil, chamados Carbonos Anoméricos. O efeito anomérico é um dos fenômenos que é classicamente explicado pela hiperconjugação. Nesses casos a hiperconjugação ocorre pela interação do par de elétrons livres do heteroátomo do ciclo com o orbital antiligante da ligação entre o carbono anomérico e seu substituinte. Com a ajuda da Química Computacional trabalhos recentes mostram que provavelmente a hiperconjugação tem pouca, ou nenhuma, contribuição para a existência desse efeito, sendo que a estabilização dos substituintes na posição axial é melhor explicada pelos efeitos das interações eletrostáticas e estéricas dos grupamentos da própria molécula. Apesar disso encontra-se na literatura evidências de que a hiperconjugação pode explicar o efeito anomérico em compostos acíclicos, como carbenos, estabilizados na posição antiperiplanar.
Modelos
Tradicionalmente, esse efeito é explicado tanto pelo conceito de estabilização do momento dipolar , como também pelo efeito de hiperconjugação, através da estabilização gerada pela interação do par de elétrons livres do heteroátomo com o orbital antiligante da ligação entre o carbono anomérico e seu substituinte . Essas explicações, especialmente a que diz respeito ao efeito de hiperconjugação, são objetos de controvérsia em publicações recentes . O modelo eletrostático mostra que a estabilidade do substituinte eletronegativo na posição axial ocorre devido à diminuição da desestabilização que ocorre quando o momento dipolar resultante entre o dipolo da ligações entre o C1 e o heteroátomo polar substituinte na posição axial e da ligação entre o heteroátomo do anel e o C1 não possui um caráter aditivo (estrutura A), como aconteceria se o substituinte estivesse na posição equatorial (estrutura B). Os dipolos alinhados causam um efeito aditivo desestabilizando a molécula e, portanto, aumentando a energia e, consequentemente, assumindo uma conformação menos estável.
Definição Moderna
Atualmente, o efeito é considerado um caso especial da preferência genérica (Efeito Anomérico Generalizado) de um substituinte no Carbono anomérico estar em posição sinclinal (gauche) em sistemas X–CH2–Y–CH2, onde X e Y são heteroátomos com pares de elétrons não-ligantes, usualmente sendo pelo menos um deles Nitrogênio, Oxigênio, Enxofre ou Flúor. Essa definição mais genérica do efeito anomérico permite a aplicação em moléculas acíclicas.
Apesar das ligações hidrogênio serem um fenômeno complexo e de muitos fatores poderem estar envolvidos na formação de complexos do tipo X-H - - - Y conectados por ligações hidrogênio, a hiperconjugação negativa n(Y) → σ∗ X-H (a qual é comumente chamada ‘componente covalente’ ou ‘componente de transferência de carga’) é um dos dois maiores efeitos estabilizadores da ligação hidrogênio, juntamente com a interação eletrostática entre dipolos . A importância de interações hiperconjugativas de um par isolado do aceptor de ligação hidrogênio com o orbital σ∗ X-H do doador é bem documentada por análise energética NBO. Como tais interações levam a um aumento na população do orbital antiligante X-H, elas alongam a ligação X-H. Quando o componente hiperconjugativo da ligação hidrogênio é fraco, o efeito de alongamento da ligação citado acima pode ser compensado por repolarização e rehibridização da ligação e então acontece a formação das chamadas ligações hidrogênio “blue-shifting” .